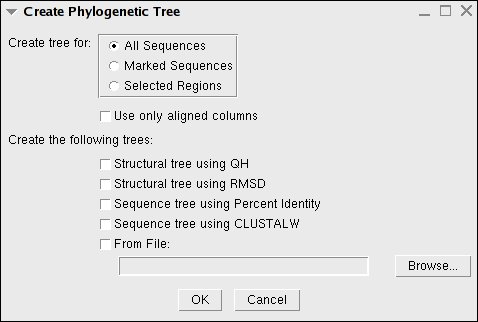
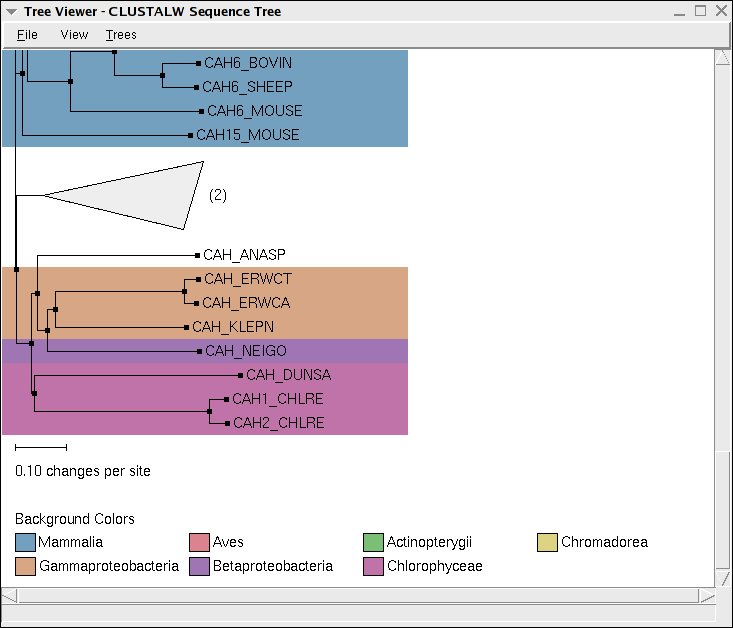
Once you have chosen which tree type to create, the Tree Viewer will be shown in simple black and white. But, you can easily use color and Tree View commands to make the data more useful (see Fig. 14).
Menu options include:
The Leaf Text option lets you choose the labels that you wish to have displayed, and you can color the labels using Leaf Color as well as the tree backgrounds using Background Color by a variety of different metrics.
You can easily collapse large parts of the tree by choosing a criteria using Collapse by, and, if you have selected a point in the tree, you can make that point the new root node of the tree using Reroot tree at selected point.