Biosynthesis and Reactivity of the Active Site of the FeFe Hydrogenases
A project supported by the U. S. National Institutes of Health
(2 R01 GM061153 F)
Principal Investigator: Thomas B. Rauchfuss
Collaborators:
David Britt, University of California, Davis
Stephen Cramer, SETI Institute
Frédéric Gloaguen, Université Bretagne Occidentale
Yvain Nicolet, Univ. Grenoble Alpes
Joshua Vura-Weis, UIUC
Giuseppe Zampella and Federica Arigoni, Universitá di Milano Bicocca
Coworkers:
Dr. Ping Wang
Dr. Xin Yu
Lauren Boedicker (jointly with Prof. Vura-Weis)
X-ray Crystallography:
Danielle Gray and Toby Woods, University of Illinois, UC
Hydrogenases are utilized by microorganisms to "process" H2, i.e. make it or oxidize it. These processes, which are important in the cellular energy management, broadly affect many redox transformations of environmental significance (e.g. sulfate reduction). Some pathogenic organisms rely on hydrogen-linked metabolic pathways, including the eubacterium Helicobacter pylori, which is responsible for gastric ulcers and cancers that affect many millions of people.
The goals of our research are to understand how these enzymes work. We pursue this question mainly by studying biomimetic models, i.e. metal complexes that look (somewhat to exactly) and behave (somewhat) like the active sites. We mainly use the techniques of organometallic chemistry. Students with interests in organometallics are well suited to contribute to this area, especially those students who like to read beyond organometallics, e.g. enzymology, "green energy", organic synthesis, and electrochemistry.
A mechanistic understanding of the hydrogenases is likely to provide the foundations for new technologies for conversions of the ultimate clean fuel, H2. Another attraction is that the catalysts are not based on platinum group metals.
[FeFe]-Hydrogenases
[FeFe]-Hydrogenases are evolutionarily more modern and in fact easier to model. Rapid progress on these sites is being made by us as well as by groups across the globe. The site consists of a diiron dithiolate with cyanide and CO as ligands as well as an appended 4Fe-4S cluster. A vacant or at least labile site is apparent on one Fe center. Substrate turn-over is localized on this site. Substrate activation is complemented by an amine-containing cofactor, which hovers over the catalytic site. Another goal of our efforts is to broaden substrate scope of the hydrogenases, such that they could be used to catalyze other kinds of reactions, such as hydrogenations, C-H bond activation, hydroformylation. A final and presently very active goal aims to elucidate the biosynthesis of the hydrogenases. In this case, we work closely with Dave Britt and his group (Lizhi Tao and Guodong Rao). Rapid progress is being made on this theme.![Structure of model for the [FeFe]-hydrogenase containing a terminal iron hydride and protonated amine cofactor](new_images/tHFe2adtNH2_101812_AtomLabels.jpg)
Structure of model for the [FeFe]-hydrogenase containing a terminal iron hydride and protonated amine cofactor
Program highlights:
- In 1999, our group crystallized models of the type [Fe2(SR)2(CO)4(CN)2]2-.
"First Generation Analogues of the Binuclear Site in the Fe-Only Hydrogenases: Fe2(SR)2(CO)4(CN)2-", Schmidt, M.; Contakes, S. M.; Rauchfuss, T. B., J. Am. Chem. Soc. 1999, 121, 9736-7. - In 2001, we discovered that diiron dithiolates catalyze the reduction of protons to H2.
"Biomimetic Proton Reduction Catalyzed by an Iron Carbonyl Thiolate", Gloaguen, F.; Lawrence, J. D.; Rauchfuss, T. B., J. Am. Chem. Soc. 2001, 123, 9476-7. - In 2001, we described the synthesis of diiron complexes containing the implied cofactor, the azadithiolate (SCH2NHCH2S), which was previously unknown.
"Diiron Azadithiolates: Synthesis, Structure, and Stereoelectronics", Lawrence, J. D.; Li, H.; Rauchfuss, T. B.; Bénard, M.; Rohmer M.-M., Angew. Chem. Int. Ed. 2001, 40, 1768-71. - In 2005, we prepared a diiron dithiolato complex containing a terminal hydride ligand.
"Characterization of the First Terminal Diferrous Hydride: Converging on the Mechanism of the Fe-only Hydrogenases" van der Vlugt, J. I.; Rauchfuss, T. B.; Whaley, C. M.; Wilson, S. R. J. Am. Chem. Soc., 2005, 127, 16012-3. - In 2007, we showed that electronically unsymmetrical diiron(I) dithiolates adopt an Hox-like structure.
"Nitrosyl Derivatives of Diiron(I) Dithiolates Mimic the Structure and Lewis Acidity of the FeFe-Hydrogenase Active Site" Olsen, M. T.; Bruschi, M.; De Gioia, L.; Rauchfuss, T. B.; Wilson, S. R. J. Am. Chem. Soc., 2008, 130, 16012-3. - In 2008, we reported that the adt cofactor participates in acid-base behavior of diiron hydrides, an essential aspect of hydrogenase action.
"Aza- and Oxadithiolates Are Probable Proton Relays in Functional Models for the [FeFe]-Hydrogenases" Barton, B. E.; Olsen, M. T.; T. B. Rauchfuss J. Am Chem, Soc. 2008, 130, 16834-16835. - In 2007-8, concurrent with studies by Marcetta Darensbourg at Texas A&M, we prepared mixed valence complexes that reproduce the rotated geometry and EPR properties of the Hox state of the enzyme. Justice, A. K.; De Gioia, L.; Nilges, M. J.; Rauchfuss, T. B.; Wilson, S. R. and Zampella, G., "Redox and Structural Properties of Mixed-Valence Models for the Active Site of the [FeFe]-Hydrogenase: Progress and Challenges", Inorg. Chem., 2008, 47, 7405-7414.
- In 2009-2011, we showed that mixed valence diiron complexes, models for Hox, activate H2 when complemented by a fast oxidant. The first direct evidence for PCET in hydrogenases. Camara, J. M. and Rauchfuss, T. B., "Mild Redox Complementation Enables H2 Activation by [FeFe]-Hydrogenase Models", J. Am. Chem. Soc., 2011, 133, 8098–8101.
- In 2012, we crystallized the ammonium-terminal hydride, key intermediate in hydrogen activation and production. Addition of one electron gives H2. The crystal was grown by Maria Carroll and the crystallography was analyzed by her father Patrick Carroll (U Penn). The diiron complex with the amine cofactor is an extremely fast catalyst for H2 production. The analogous propanedithiolate is a poor catalyst.
M. E. Carroll, B. E. Barton, T. B. Rauchfuss and P. J. Carroll, "Synthetic Models for the Active Site of the [FeFe]-Hydrogenase: Catalytic Proton Reduction and the Structure of the Doubly Protonated Intermediate", J. Am. Chem. Soc. 2012, 134, 18843-18852.
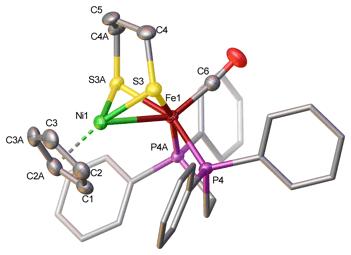
- In 2013 we crystalized a rotated reduced diiron dithiolate, Fe2(2,2-Et2pdt)(CO)4(dppv). Preparation by Dr. Wenguang Wang. Crystallography by Curtis Moore and Arnold Rheingold (UCSD).
- 2015:
Ryan Gilbert-Wilson develops a route to Fe(0) from 57Fe and collaborates with the Lubitz group to label the diiron subsite of Hyd-A1 in Chlamydomonas reinhardtii using the Grenoble Insertion method.
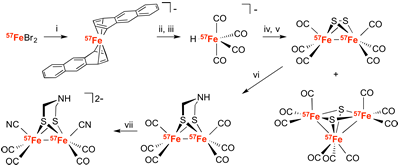
"Spectroscopic investigations of [FeFe] hydrogenase maturated with [57Fe2(adt)(CN)2(CO)4]2-" Ryan Gilbert-Wilson, Judith F. Siebel, Agnieszka Adamska-Venkatesh, Cindy C. Pham, Edward Reijerse, Hongxin Wang, Stephen P. Cramer, Wolfgang Lubitz, Thomas B. Rauchfuss.

Terminal hydride states are observed by NRVS using our 57Fe labeling methods. Pelmenschikov, Birrell, Pham, Mishra, Wang, Sommer, Reijerse, Richers, Tamasaku, Yoda, Rauchfuss, Lubitz, Cramer, Reaction Coordinate Leading to H2 Production in [FeFe]-Hydrogenase Identified by Nuclear Resonance Vibrational Spectroscopy and Density Functional Theory. J. Am. Chem. Soc. 2017, 139, 16894-16902. Reijerse, Pham, Pelmenschikov, Gilbert-Wilson, Adamska-Venkatesh, Siebel, Gee, Yoda, Tamasaku, Lubitz, Rauchfuss, Cramer, Direct Observation of an Iron-Bound Terminal Hydride in [FeFe]-Hydrogenase by Nuclear Resonance Vibrational Spectroscopy. J. Am. Chem. Soc. 2017, 139, 4306-4309.
Cass Richers develops a route to the diruthenium azadithiolate with which Hyd-A1 in Chlamydomonas reinhardtii was again modified. We learned a lot:
Maturation using [Ru2(ADT)(CO)4(CN)2]2- and [(µ-H)Ru2(ADT)(CO)4(CN)2]- gave the same terminal hydride.
The redox potentials for the [4Fe4S]2+/+ couple differs for HydA1-[Ru(II)Ru(II)]H-ADT vs HydA1-[Ru(I)Ru(I)]H-ADT. The Ru(II) species reduces at -270 mV whereas the [Ru(I)Ru(I)] state reduces at -370 mV. This difference signifies the strong electronic contact between the two sub-clusters.
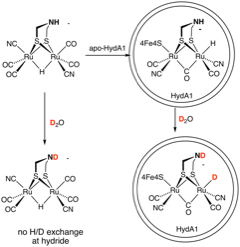
The hydride in HydA1-[2Ru]H-ADT fully H/D exchanges with solvent, whereas the hydride in HydA1-[2Ru]H-PDT does not. This result demonstrates the mechanistic role of the adt.
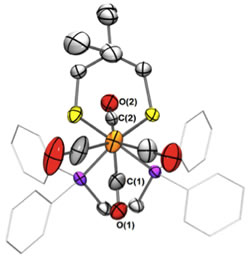
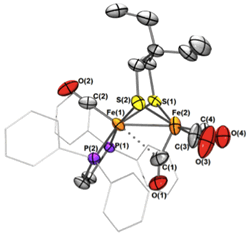
We make Ru2(ADT)(CO)6 (see pic) and [(µ-H)Ru2(ADT)(CO)4(CN)2]2-. The Max Planck group conducted maturation. The hydride shifts to terminal position. It exchanges with D2O. The corresponding PDT derivatives also gives terminal hydride, but it does not exchange with D2O.
(CO)6.jpg)
- 2019
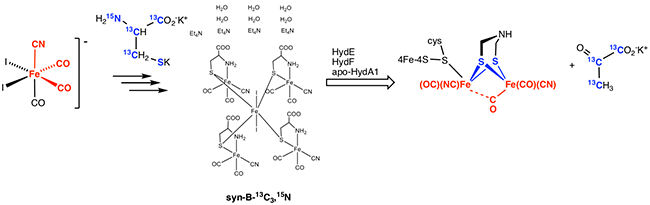
With the Britt group, we show that cysteine is the source of sulfur for ADT, but not the source of CH2 or NH. This insight is provided by the use of "syn-B" a meta-stable synthetic analogue of "Complex B". Syn-B is a carrier for the Fe(cys)(CN)(CO)2 module (as is Complex B). Also evident from these experiments is that the carrier delivers S-Fe-CN-(CO)2 intact and without scission of Fe-C or Fe-S bonds.
- 2022
With the Britt group again, we show that [Fe2(SH)2(CN)2(CO)4]2- allows the biosynthesis of active hydrogenase without HydG and HydE. A significant advance in the area.
- 2023
With the Britt group again, we will show that ....the biosynthesis ... stay tuned...
We finally reinvestigated [Fe2[(SeCH2)2NH)](CN)2(CO)4]2-, which was claimed many years ago based on very strange data. This species is the analogue of the natural (SCH2)2NH complex. Dr. Xin Yu learned how to make it and figured out why others failed.

Dr. Xin Yu, expert on Fe2(ER)2L6 (E = S, Se).
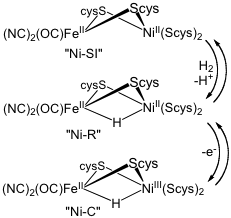
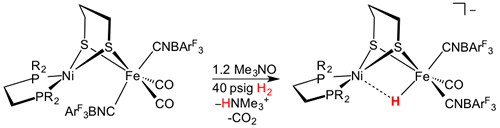
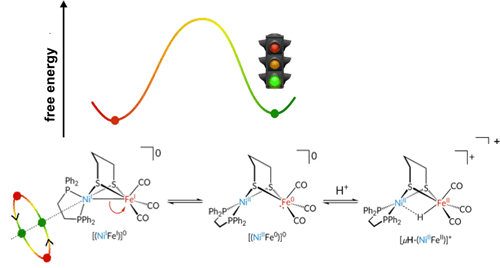
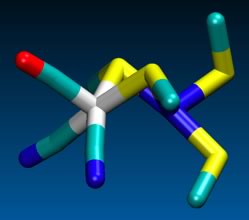 [NiFe]-Hydrogenases are more pervasive than [FeFe] hydrogenases. Unlike the FeFe] enzymes, the [NiFe] site binds the hydride between the two metal sites, Fe(CO)(CN)2 and Ni(Scys)2 centers linked by a pair of cysteinyl thiolates. The ensemble provides a redox-active receptor for protons.
[NiFe]-Hydrogenases are more pervasive than [FeFe] hydrogenases. Unlike the FeFe] enzymes, the [NiFe] site binds the hydride between the two metal sites, Fe(CO)(CN)2 and Ni(Scys)2 centers linked by a pair of cysteinyl thiolates. The ensemble provides a redox-active receptor for protons.