Lattice Microbes and pyLM

Lattice Microbes is a software package for efficiently sampling trajectories from the chemical and reaction-diffusion master equations (CME/RDME) on high performance computing (HPC) infrastructure using both exact and approximate methods. Along with it, the pyLM problem solving environment allows for customizable simulations.
For more information, see the Lattice Microbes/pyLM home page.
Representative papers related to Lattice Microbes/pyLM:
Elijah Roberts, John E Stone, and Zaida Luthey-Schulten.
Lattice Microbes: high-performance stochastic simulations of the reaction-diffusion master equation
J. Comput. Chem., 34(3):245-55, 2013
E. Roberts, A. Magis, J.O. Ortiz, W. Baumeister, and Z. Luthey-Schulten
Noise Contributions in an Inducible Genetic Switch: A Whole-Cell Simulation Study
PLoS. Comput. Biol., 7(3):e1002010, 2011
E. Roberts, J.E. Stone, L. Sepulveda, W.W. Hwu, and Z. Luthey-Schulten
Long time-scale simulations of in vivo diffusion using GPU hardware
In Proceedings of the 2009 IEEE International Symposium on Parallel & Distributed Processing, 2009
populationFBA Example Script and Data
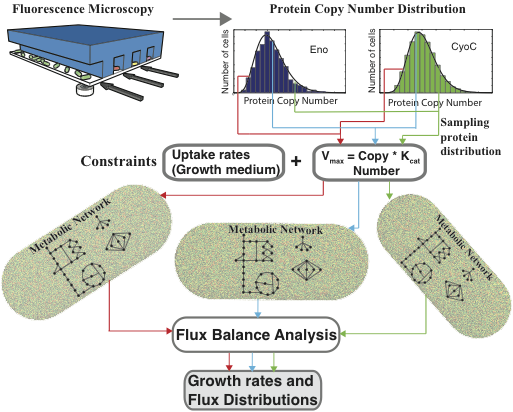
populationFBA includes a Matlab script and data file which, when run, samples the copy number distributions of over 350 metabolic proteins in E. coli, and uses them to set constraints on reaction fluxes throughout a comprehensive model of metabolism. A detailed description of this methodology can be found in:
P. Labhsetwar, J.A. Cole, E. Roberts, N.D. Price, Z. Luthey-Schulten. Heterogeneity in protein expression induces metabolic variability in a modeled Escherichia coli population. Proc. Nat. Acad. Sci., 110(34):14006-11, 2013, doi: 10.1073/pnas.1222569110
Please download both the script (populationFBA_ZLS.m) and data (populationFBA_ZLS_Data.mat) files:
http://opencobra.sourceforge.net/openCOBRA/Welcome.html
continuumColony for 3DdFBA colony simulations
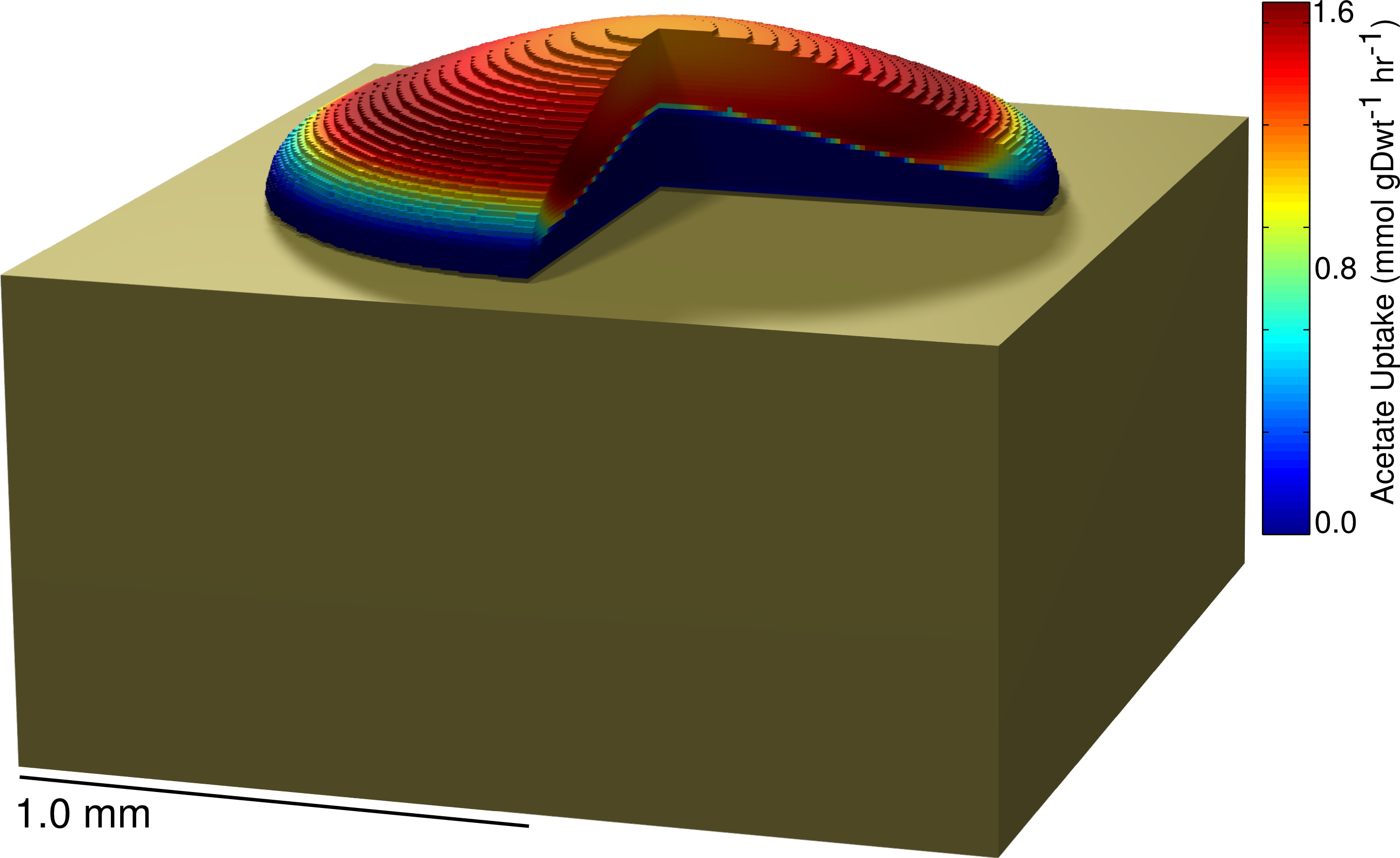
continuumColony Includes a compressed directory containing source code for simulating the growth of a colony using the 3DdFBA methodology described in:
Cole JA, Kohler L, Hedhli J, and Luthey-Schulten Z. (2015) Spatially-Resolved Metabolic Cooperativity Within Dense Bacterial Colonies. BMC Syst. Biol. 2015, 9(15), doi:10.1186/s12918-015-0155-1
This download also contains useful MATLAB files for generating input FBA tables and viewing simulation data, as well as a user guide.
https://opencobra.github.io
Dynamical Network Analysis
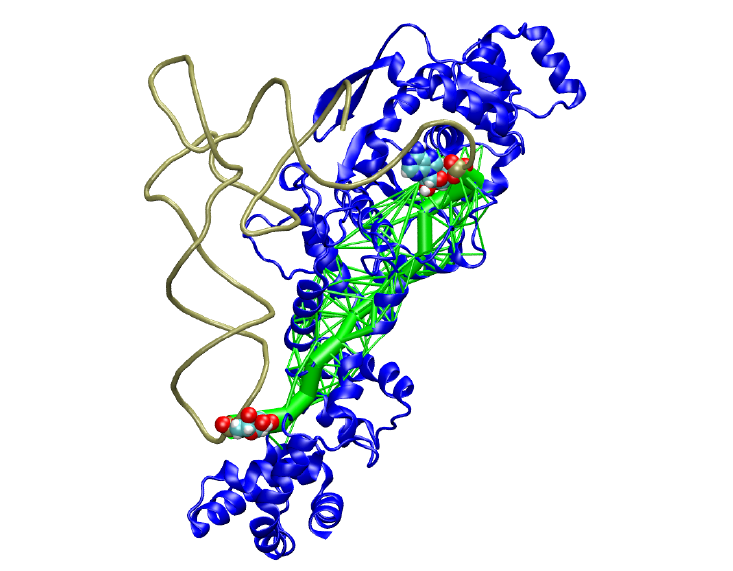
Dynamical Network Analysis is a method of characterizing allosteric signalling through biomolecular complexes.
For more information, see the Dynamical Network Analysis home page.
Representative papers related to Dynamical Network Analysis:
R.W. Alexander, J. Eargle, and Z. Luthey-Schulten
Experimental and computational analysis of tRNA dynamics
FEBS Letters, 584(2):376-386, 2010
Alexis Black Pyrkosz, John Eargle, Anurag Sethi, and Zaida Luthey-Schulten
Exit strategies for charged tRNA from GluRS
J. Mol. Biol., 397:1350-1371, 2010
Anurag Sethi, John Eargle, Alexis Black, and Zaida Luthey-Schulten.
Dynamical Networks in tRNA:protein complexes
Proc. Natl. Acad. Sci. U S A, 106(16):6620-6625, 2009
Supplementary Material
MultiSeq 2.0

MultiSeq is an extension to VMD that provides a combined sequence and structure bioinformatics analysis environment. VMD and MultiSeq are available for many platforms, including Windows, Mac OS X, Linux, and Solaris. They can be downloaded from the NIH Resource for Macromolecular Modeling and Bioinformatics. Tutorials are also available for learning how to use MultiSeq and VMD.
For more information, see the MultiSeq home page.
Representative papers related to MultiSeq:
E. Roberts, J. Eargle, D. Wright, and Z. Luthey-Schulten. MultiSeq: Unifying sequence and structure data for evolutionary analysis. BMC Bioinformatics, 7:382, 2006.
J. Eargle, D. Wright, and Z. Luthey-Schulten. Multiple Alignment of protein structures and sequences for VMD. Bioinformatics, 22:504, 2006.
SeqQR 1.0.1
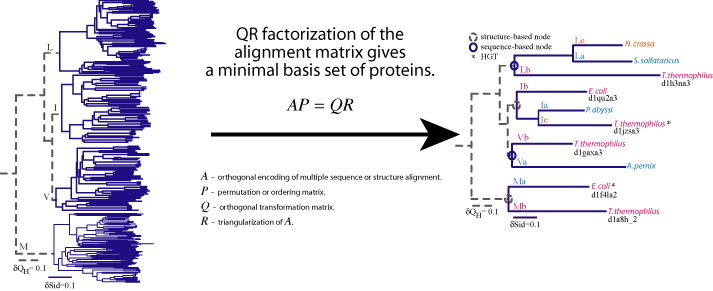
Sequence QR (SeqQR) is a program that generates a nonredundant set of proteins that represents the topology of the molecular phylogenetic tree of the homologous group.
SeqQR is available on the following platforms:
Representative papers related to SeqQR:
A. Sethi, P. O'Donoghue, and Z. Luthey-Schulten. Evolutionary profiles from the QR factorization of multiple sequence alignments. PNAS, 102:4045, 2005.
P. O'Donoghue and Z. Luthey-Schulten, Evolutionary profiles derived from the QR factorization of multiple structural alignments gives an economy of information. J. Mol. Biol., 346:875, 2005.
P. O'Donoghue, and Z. Luthey-Schulten. On the evolution of structure in aminoacyl-tRNA synthetases. Microbiol. Mol. Biol. Rev., 67:550, 2003.
Tessellator 1.0
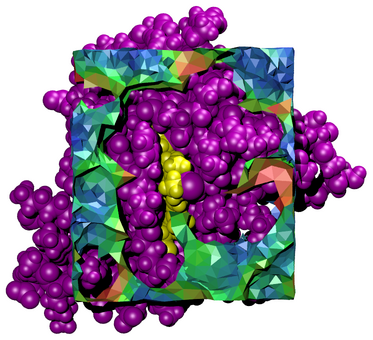
Tessellator is an application for the tessellation of 3D volume in and around biological molecules. It creates Tcl script files which are run within VMD to visualize volumes in context with their associated molecular structures.
For help with installation and use of Tessellator, click here.
For supplementary material associated with the paper "Visualizing the Dual Space of Biological Molecules", click here.
Tessellator is available on the following platforms:
Representative paper related to Tessellator:
J Eargle, and Z. Luthey-Schulten, Visualizing the Dual Space of Biological Molecules. Comp. Biol. Chem., 30:219, 2006.
Ionize 1.6.0
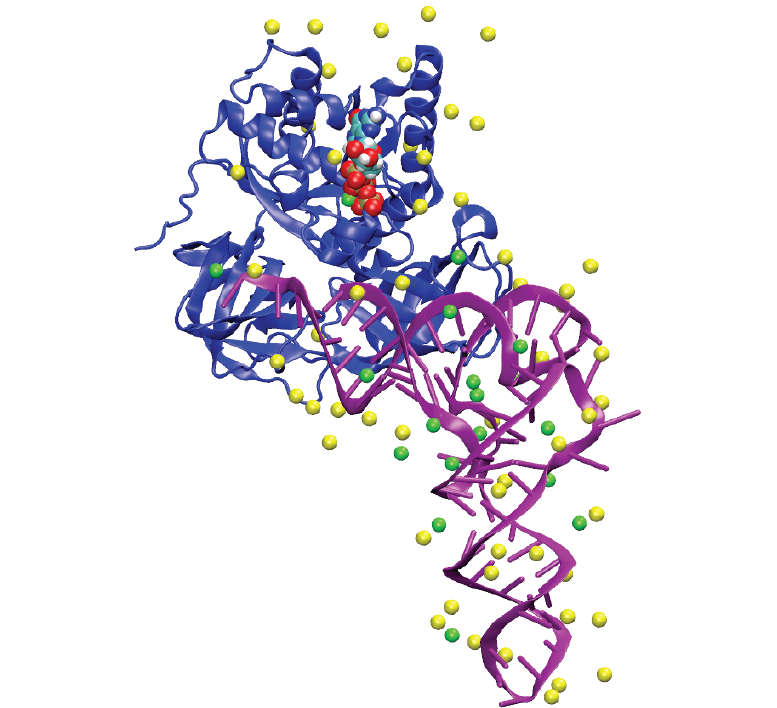
Ionize is an application for the placement atomic ions around 3D biomolecular structures. It uses a pdb file with associated atomic charges to create a grid of electrostatic potential around the structures. An ion is placed at the lowest energy gridpoint, and the grid is then recalculated for the placement of the next ion. Multiple ion types can be placed in a specified order.
For help with installation and use of Ionize, click here.
Ionize is available as source code:





